амиды карбоновых кислот
АМИДЫ КАРБОНОВЫХ КИСЛОТ [от ам(миак)]
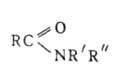
ацилпроизводные аммиака или аминов, соед. общей формулы RC(O)NR'R". Незамещенные у атома N амиды (А.) RCONH2 наз. первичными; моно- и дизамещенные А. RCONHR' и RCONR'R" (R' и R"-opr. остаток) — соотв. вторичными и третичными. Соед., содержащие две ацильные группы у атома азота RCON(R')COR", наз. имидами, а соединения с тремя ацильными группами RCON(COR')COR" — триациламинами. По др. классификации соед., содержащие одну, две или три ацильные группы, наз. соотв. первичными, вторичными или третичными. Циклич. аналогами А. являются лактамы. Об А. сульфокислот см. сульфамиды.
Названия первичных А. производят от названий соответствующих кислот, напр. НСОNH2-формамид, или амид муравьиной кислоты, CH3СОNH2-ацетамид, или амид уксусной кислоты, C6H5СОNH2 — бензамид, или амид бензойной кислоты. В названиях N-замещенных А. заместители перечисляются перед названием незамещенного А., напр. HCON(CH3)2 — N.N-диметилформамид.
Кроме жидких формамида и N-метил-формамида, первичные и вторичные А. — кристаллич. вещества, большинство третичных-жидкости. Низшие алифатич. А. хорошо раств. в воде, простейшие ароматические — умеренно в горячей воде. Между молекулами А., содержащими хотя бы один атом Н при атоме N, возникают водородные связи.
В ИК-спектрах первичных А. имеются две полосы поглощения, характерные для своб. NH2 — группы, ок. 3500 и 3400 см−1 и две полосы, характерные для группы C=O, при 1690–1630 см−1 (т. н. амкдная полоса I) и 1620–1590 см−1 (амидная полоса II). Вторичные А. имеют одну полосу поглощения NH-группы в области 3460–3420 см−1 и две полосы поглощения группы C=O в областях 1690–1630 и 1550–1510 см−1. У третичных А. — одна полоса поглощения группы C=O ("амид I") в области 1670–1630 см−1. В спектре ЯМР сигналы протонов NH2-группы проявляются в интервале от 5 до 8 м. д. в шкале
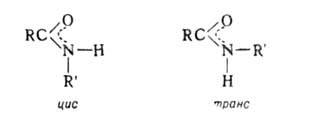
Вследствие частичной двоесвязанности N=C и, следовательно, затруднения своб. вращения вокруг связи C(O)—N А. могут существовать в цис- и транс-формах:
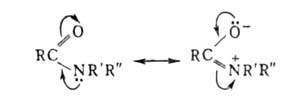
В водных растворах А. обычно имеют нейтральную реакцию, что обусловлено сопряжением своб. электронной пары атома N с двойной связью карбонильной группы:
Однако первичные и вторичные А. могут проявлять слабые амфотерные свойства, а третичные — слабые основные. Так, с сильными минеральными кислотами они образуют непрочные легко гидролизующиеся соли. Основные свойства N-алкилзаме-щенных А. выражены сильнее, чем у незамещенных. Например, N,N-диметилацетамид образует с HCl соль, устойчивую в виде конц. водных растворов, а с HClO4 и H2PtCl6 — прочные хорошо кристаллизующиеся соли. В среде уксусного ангидрида А. количественно оттитровываются раствором HClO4. При взаимодействии с щелочными металлами у первичных и вторичных А. атом Н аминогруппы замещается металлом, образуя, напр., RCONHNa.
При кипячении с конц. водными растворами минеральных кислот или щелочей А. гидролизуются до кислот. Под действием HNO2 легко дезаминируются: RCONH2 + HNO2 → RCOOH + N2 + H2O. Восстанавливаются до аминов RCH2NR'R" натрием в спиртовой среде, алюмогидридом Li или H2 — над меднохромовыми катализаторами. Первичные А. дегидратируются до нитрилов под действием P2O5, Al2O3, SiO2, H3PO4 или др.; при действии гипобромитов или гипохлоритов в щелочном растворе превращаются в первичные амины (Гофмана перегруппировка). Взаимод. вторичных и третичных А. с PCl5, SOCl2 и т. п. приводит к имидоилхлоридам RCC1=NR' или α-хлориммониевым солям RCC1—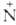 R'R"C1−, которые при нагревании расщепляют- , ся на нитрилы RCN и алкилгалогениды (см. Брауна реакция).
R'R"C1−, которые при нагревании расщепляют- , ся на нитрилы RCN и алкилгалогениды (см. Брауна реакция).
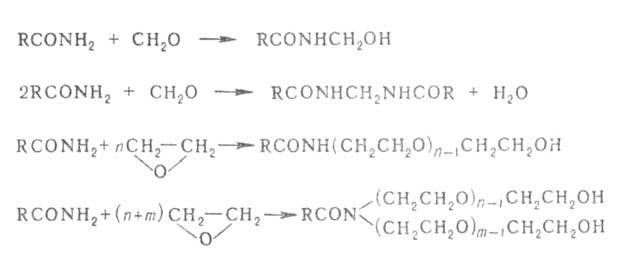
С бромом и хлором А. образуют соотв. N-бром- и N-хлорамиды, с формальдегидом и окисью этилена-N-метилоламиды, N,N'-метилен-бис-ациламиды и разл. N-оксиэтильные производные амидов, имеющие большое пром. значение, напр.:
В промышленности А. синтезируют взаимод. кислот или чаще их хлорангидридов, ангидридов, эфиров с NH3 (аммонолиз) либо амином (аминолиз). Еще один пром. способ — неполный гидролиз нитрилов в присуг. H2SO4 или Cu: RCN + H2O → RCONH2. Разработаны непрерывные контактно-каталитич. аммонолиз и аминолиз кислот при 200–280 С в присутствии катализаторов дегидратации (Al2O3, SiO2 и др.); выход А. 95–98%.
В'лаб. условиях А. можно синтезировать также реакцией кетенов с NH3 или амином (напр., CH2=C=O + NH3 → CH3CONH2), N-алкилзамещенные А. — взаимод. амидов с алкилгалогенидами, N-алкил- и N-арилзамещенные — с использованием Бекмана перегруппировки или перегруппировки Шмидта:
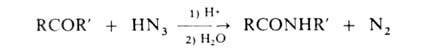
Образование А. используют для защиты аминогруппы и для идентификации первичных и вторичных аминов (преим. в виде ацетамидов и бензамидов), а также карбоновых кислот (в виде незамещенных А., анилидов, бензиламидов). Особое значение методы защиты МH2 — группы имеют в синтезе пептидов (см. белки).
А. — пластификаторы бумаги, искусственной кожи, ПВХ, экстрагенты некоторых радиоактивных металлов, сырье в производстве полимеров, промежут. продукты в синтезе красителей и сульфамидных препаратов и др.
Лит.: Бюлер К., Пирсон Д., Органические синтезы, пер. с англ., ч. 2, М.. 1973, с. 384–430; Органикум, пер. с нем., т. 2, М., 1979, с. 84–99; Общая органическая химия, пер. с англ., т. 4, М., 1983, с. 388–536; Kirk-Othmer encyclopedia, 3 ed., v. 2, N.Y.-[a.o.], 1978, p. 252–59.
Н. К. Садовая
